催化审稿人建议加计算,怎么加?别担心,审稿人想看的计算全都有!
2023-01-18
admin 53 0 核心提示:最近收到很多类似的反馈,催化类文章审稿人要求用DFT计算对某个科学问题进行具体解释。与其在修稿期间匆匆忙忙加计算,不如从
最近收到很多类似的反馈,催化类文章审稿人要求用DFT计算对某个科学问题进行具体解释。与其在修稿期间匆匆忙忙加计算,不如从一开始构思时就采用DFT计算与高端表征相结合的思路,这其实是
顶刊标配做法 。
但是催化类的DFT计算往往较为复杂,大家担心短时间内学不会。Materials Studio是入门DFT计算的首选软件,其基于Windows界面的强大建模、结果分析功能,可
将学习门槛降到最低 。
并且,杨站长通过自己开发的脚本、制作的表格已将自由能台阶图、d带中心、过渡态、电荷差分密度分析等手段固定化、流程化,
让催化计算进一步简化。
华算科技开办的
Materials Studio光电热催化培训,由全职技术专家杨老师主讲,精心挑选案例,紧跟科研热点!
十多年DFT计算建模实战经验、内功心法,倾囊相授!
不限时间不限地点,随时学习!
无限次回放、永久答疑!
本次课程基于CASTEP、
DMol
3
模块设计,直击催化科研热点,
通过28小时高强度实操培训,用通俗的语言将HER/OER/ORR/CO
2
RR台阶图、火山图、d带理论、过渡态、带隙工程、能带电位匹配等催化计算核心问题讲透彻,使大家能将DFT计算用到自己的文章中。
报名方式:识别下方二维码报名
,或者联系手机181-2638-7652。
注:此课程为MS催化专题课程,配合零基础培训食用更佳!(点击链接跳转)
通过华算科技三年多的培训,已有超过1000名学员完成了系统的DFT催化计算课程,大量学员学以致用,顺利发表文章。在通往顶刊的路上,他们早已快人一步!
培训采用线上直播授课形式,为了保证学习效果,提供无限回看,课后继续提供超算账号, 建立永不解散的课程群,永久答疑! 现在报名,买一送三,赠送下面三套总价2497元的计算课程,让你计算技能更加全面。
杨站长:
华算科技全职技术资深专家,深圳市孔雀计划海外高层次人才。曾就职于德国马克思普朗克研究所,日本WPI研究所,并曾在芬兰阿尔托大学进行长期访问,作为PI主持欧盟、日本科研项目6项。
拥有13年以上Materials Studio软件使用经验,主要从事固态相变的第一性原理研究、电化学固液界面的AIMD研究与超分子化学中的分子动力学模拟。
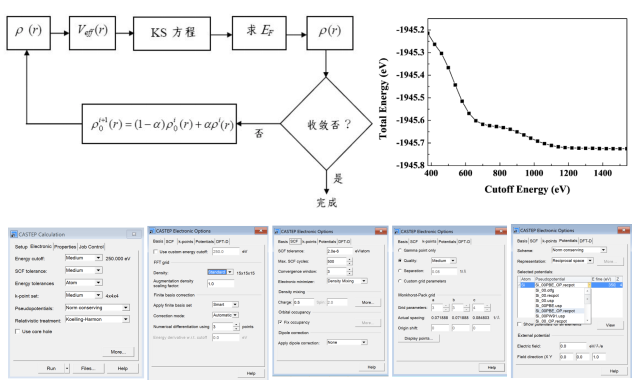
1. 波恩-奥本海默近似,Hatree-Fock近似, 2. 密度泛函理论,Kohn-Sham方程,交换关联势,LDA/GGA,电子自洽循环
MS在Windows及Linux系统中的合规安装与运行脚本(实操
)
3. Materials Studio在Linux上的安装
用于发表文章的数据,大部分是用Linux服务器计算得到。学完这部分知识,能够自主实现MS在服务器上的安装,并掌握在有无作业管理系统时,作业脚本的编写与运行方法。
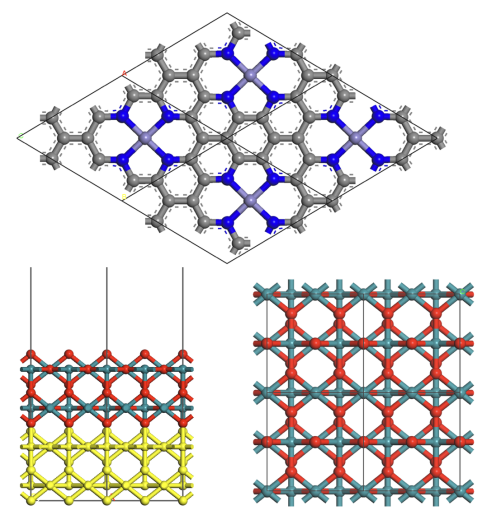
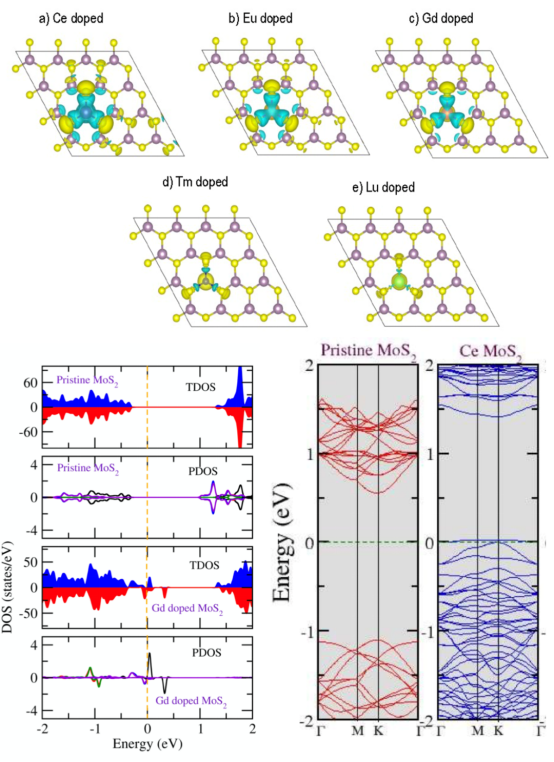
3. 掺杂石墨烯与二维层状结构MoS
2
, MXene搭建
从免费数据库中的体块结构入手,手把手带大家进行经典文献中模型的重现。本章带大家进行表面、单层结构催化剂的建模操作,全面讲解构建催化剂表面时所须注意的细节问题。
使所建模型既“合理”又“可算”,在满足高水准文章要求的同时也兼顾计算量。所设计案例的模型分为三类—金属表面,金属氧化物表面与单层材料,同时讲解掺杂方法,基本覆盖所有催化剂类型。
本章将带大家了解电、热、光催化的基本原理及其催化剂所起作用的不同。同时也将介绍反应路径、中间态与过渡态的物理意义,光生载流子、带边等基本概念;电化学中如何通过改变电极电位可调控反应的热力学,怎样为化学反应研究打开了一个新的维度。
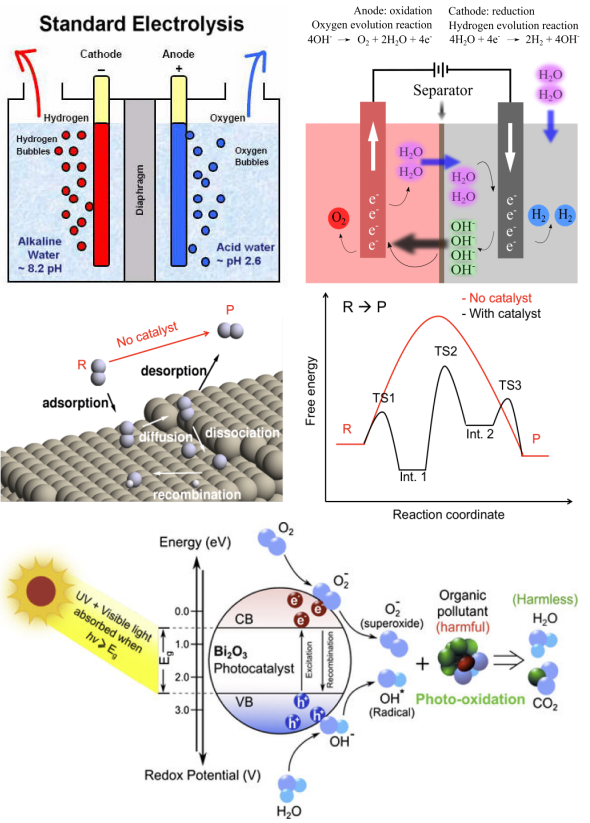
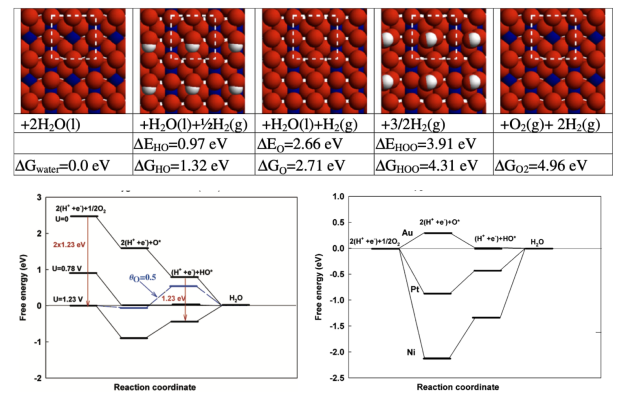
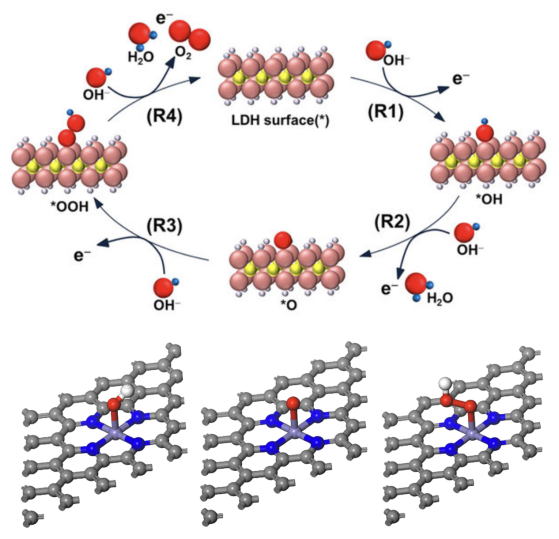
4. 台阶图的解读,中间体的自由能之差与热力学过电位 电化学电位能够强制打破反应物与生成物的热力学平衡,其机理可以简化成水分子间质子的跃迁。
Norskov提出的计算氢电极模型CHE被广泛采用,本章将为大家讲解其中所涉及的基本思想及其与真实电化学之间的差异。此外,本章将介绍能够减少计算量的吉布斯自由能矫正脚本的编写思路及使用方法。
H
ER电催化反应热力学—自由能台阶曲线(建模+计算实操)
通过最简单的操作快速实现HER催化反应中H*、分子吸附与解离吸附H
2
O模型的总能计算与零点振动能、热力学校正的计算。
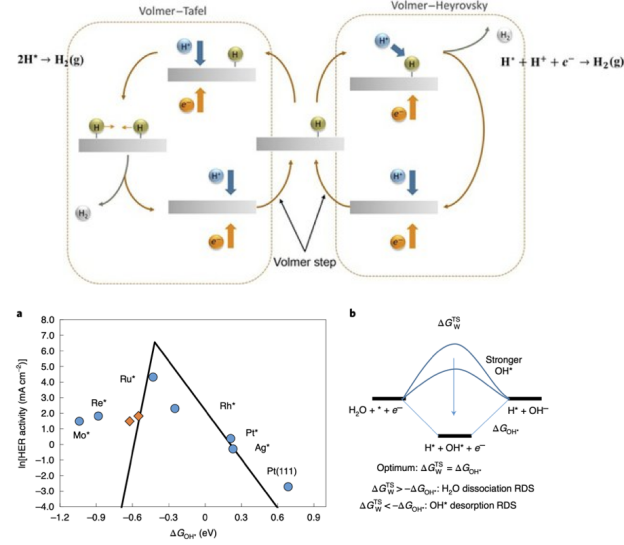
使大家了解Volmer-Tafel与Volmer-Heyrovsky过程的差异,以及酸碱条件下HER吉布斯自由能变化的差异。
OER/ORR电催化自由能台阶曲线-- 金属氧化物,
DMol
3
模块
1. 金属氧化物表面上OER/ORR反应中间体吸附构型的搭建 前面理论部分描述了怎样求解台阶图的方法论,但实际操作并不复杂。在给出脚本的前提下,只需要将14个数输进给定的表格中即可出图。 本章以金属氧化物表面催化剂为例,为大家介绍OER/ORR反应多步台阶图中的自由能该如何计算,以及不同电压对自由能的影响。
OER/O
RR电催化自由能台阶曲线— 单原子催化剂,CASTEP模块
1. 单原子催化剂上OER/ORR反应中间体吸附构型的搭建 3. CASTEP计算部分原子振动对自由能矫正贡献的方法 本章为大家介绍单原子催化剂OER/ORR反应多步台阶图中的自由能该如何计算,以及不同电压对自由能的影响。
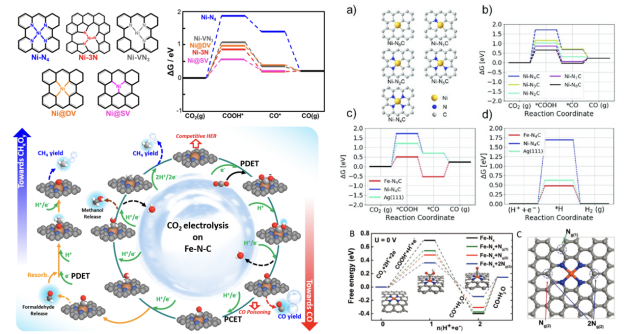
CO
2
RR电催化反应热力学—自由能台阶曲线(建模+计算实操)
1. 二维单层材料上CO
2
RR反应中间体吸附构型的搭建
本章以二维单层材料催化剂为例,为大家介绍CO
2
RR反应中的各个步骤、不同转移电子数对应的产物与决速步,与HER的竞争,带大家进行台阶图中中间态结构的构建与自由能计算。同时将讲解以MXene等为催化剂的甲酸还原路径建模方法。
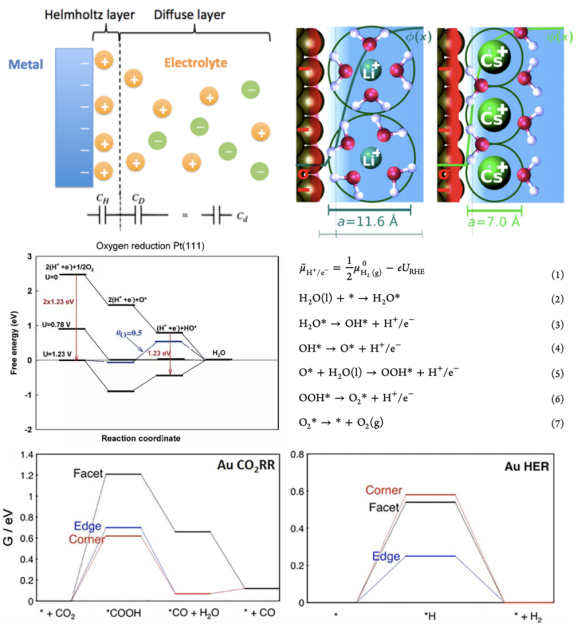
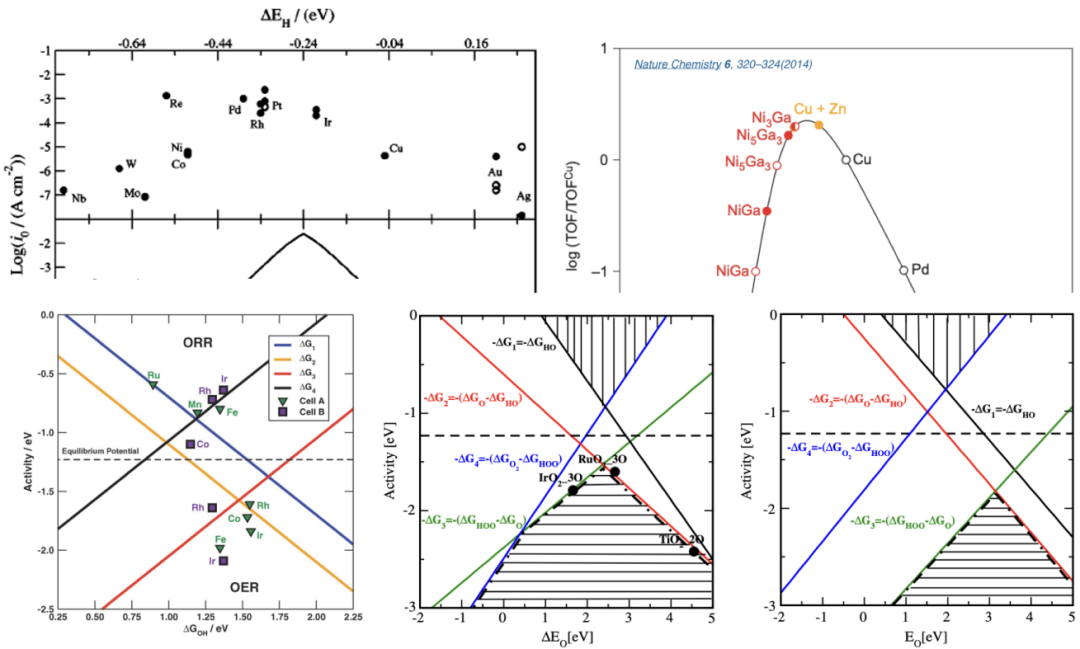
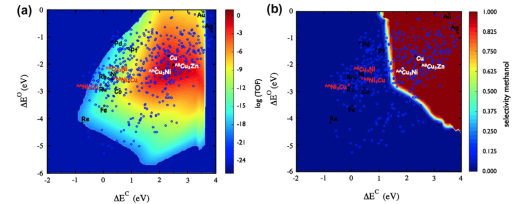
2. neither too strong nor too weak 3. 针对不同种类、不同合金比例、不同晶面的催化剂的ORR与HER火山曲线的解读 4. BEP关系与Micro-kinetic Model 电流或电位的火山图包含了多种催化剂表面活性的线性比例关系,提供了最佳催化剂设计标准,同时也为实验与理论计算搭起了桥梁。
本章将带大家了解火山图的意义,催化活性与吸附能之间的关系,以及怎样从施加偏压着手来计算OER/ORR的火山图。此外,也将为大家讲解不同催化剂反应热力学与动力学之间的线性关系,与微动力学火山图。
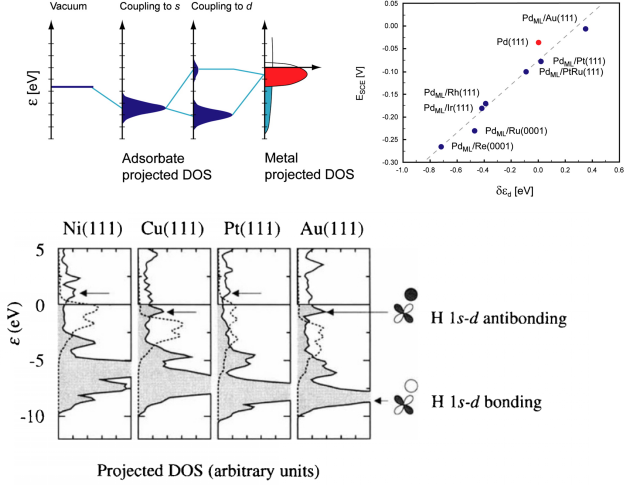
d带理论在催化性质分析中扮演着重要的角色。本章将以通俗的语言为大家介绍该理论的核心及应用价值,并带大家进行d带中心计算实操。
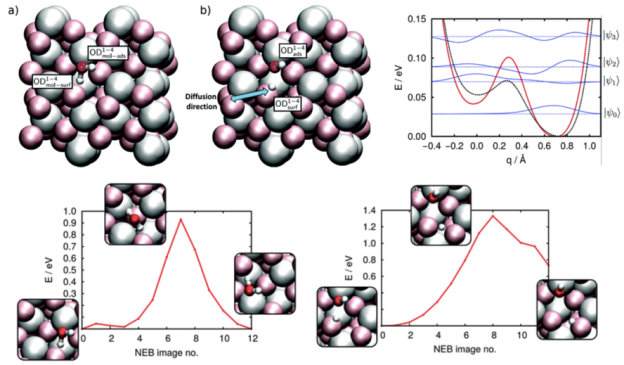
反应能否发生决定于热力学,但发生的快慢决定于动力学。本章将使大家掌握化学反应中过渡态搜索算法中的基本概念,以及激活能的计算方法,补齐自由能曲线各中间态之间的“缺失的弧线”。
本章为大家介绍CO
2
还原成不同产物的全部路径,并带大家进行CO
2
分子在表面的吸附构型及还原过程中间态的构建,以及不同吸附构型分解势垒的计算。同时引进MS全新模块FlexTS进行全势能面搜索,以确认最小能量反应路径。
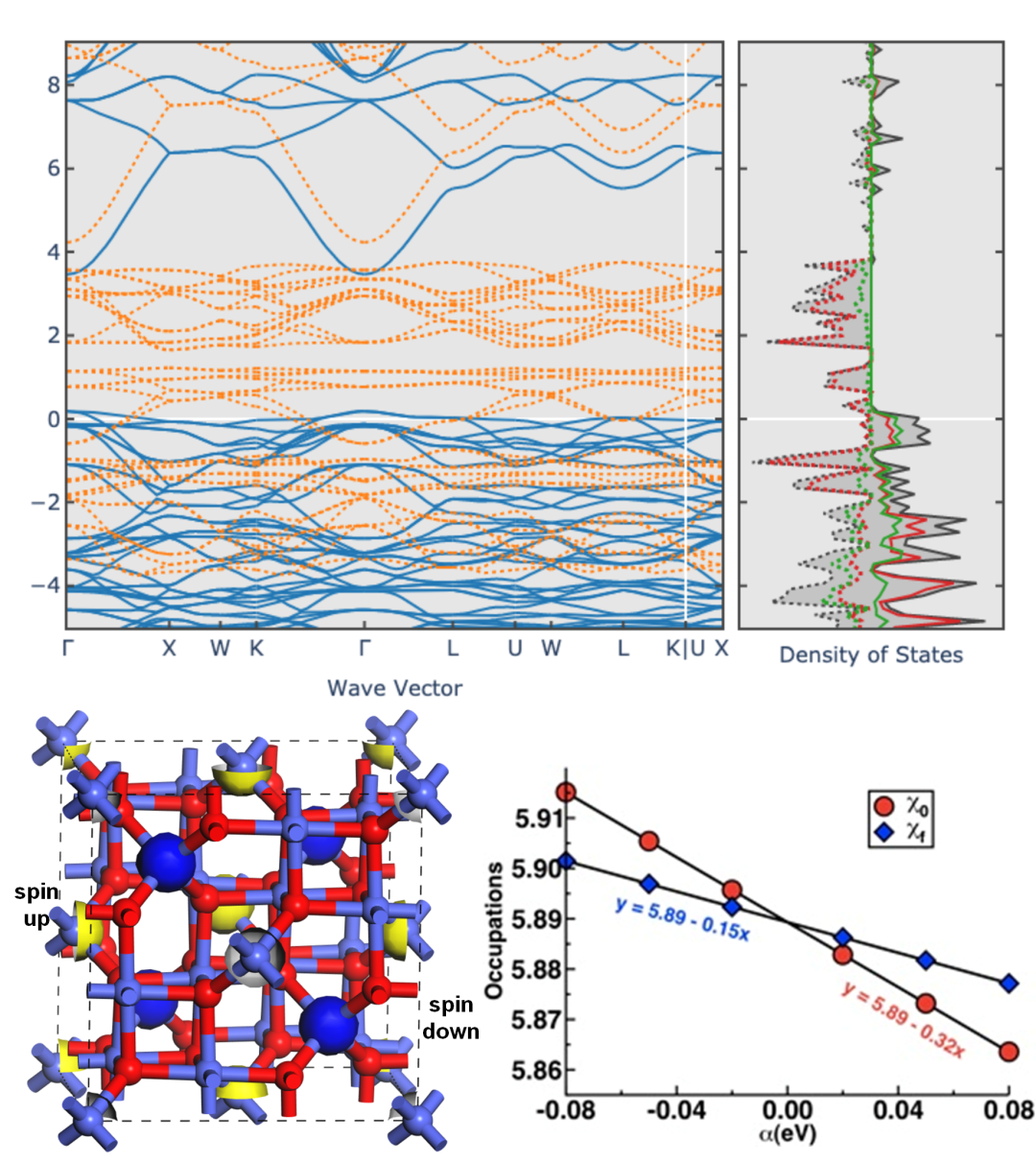
带隙低估是普通DFT应用在半导体光催化领域时最大的问题。采用杂化泛函,可得到与实验值更符合的带隙。带有自旋极化的体系以及序磁性体系中,原子的磁矩设置不但影响体系的总能,对催化剂的性能也有很大影响。
DFT+U修正了强关联体系能量的同时,也会带来副效应--在一定程度上增加带隙值。本章将为大家讲解这些方法的原理及操作过程。
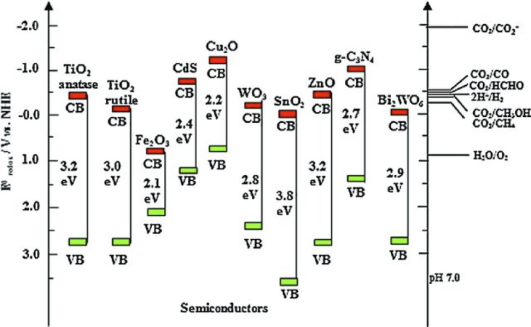
本章为大家讲解半导体光催化领域的历史及进展,带隙工程,掺杂调节能带结构的深层机制,掺杂及缺陷体系的形成能计算方法,失主受主对间的电荷补偿机制,电子-空穴的有效分离方法。并从分波态密度上分析原因,从而拔高文章档次。
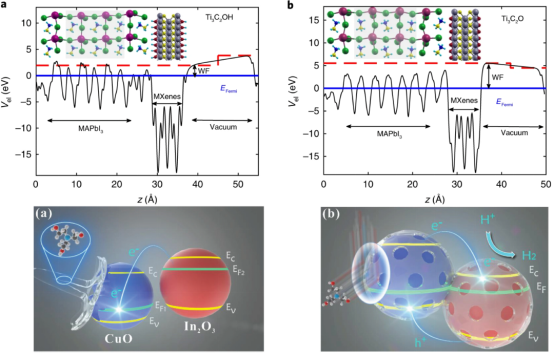
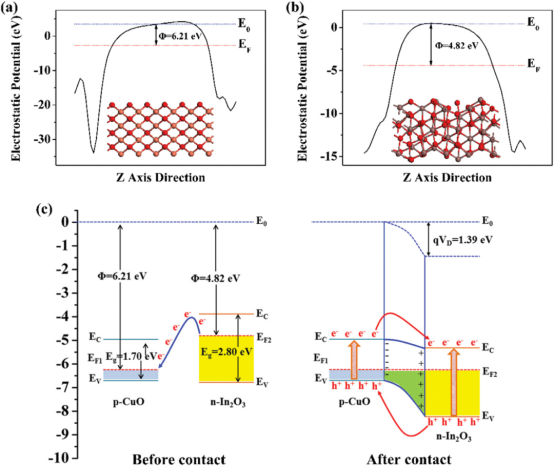
本章带大家掌握表面功函数的计算,以及通过功函数与能带结构进行能带电位匹配,探讨异质结构如何促进光生电子、空穴的复合。并对半导体材料异质结构进行建模,考察界面间电荷转移,分析电子的积累与耗尽,以及界面内建电场的方向与电势差。
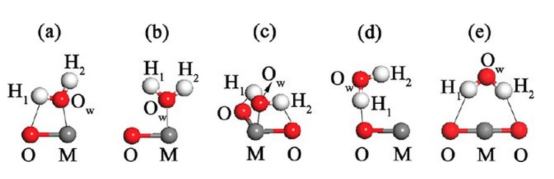
本章将为大家介绍如何构建吸附模型。搜寻最稳定的分子吸附构型,需要在复杂的的势能面上找到全局最低点,计算量较大。采用一定的经验,可以帮助减小计算的个数。另外采用MS的分子力学模块可以自动搜寻复杂分子的吸附构象,只是其应用有一定的局限性。
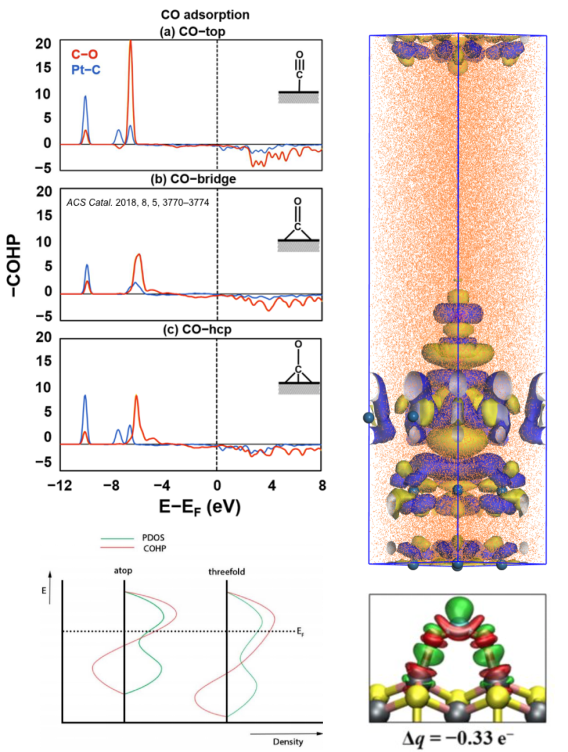
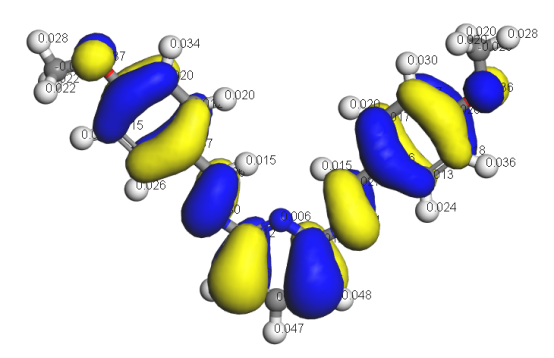
吸附与活化的关键在于分析化学键,而化学键根源于电子性质,常规电子性质分析对化学反应十分重要,本章将从电子态密度、晶体轨道哈密顿布居、差分电荷密度、电荷与键的布居等角度使大家掌握如何从电子性质上考量化学键的形成、断裂、削弱的内在原因。
主办单位: 深圳华算科技有限公司(拥有VASP、Materials Studio、Gaussian、LAMMPS商业版权)
深圳浦华系统技术有限公司(Materials Studio官方代理)
培训形式: 录播课程,提供回放视频,课程群永不解散,随时提问,及时解答。
课程采用Windows版本Materials Studio建模+参数设置,Linux版本完成计算任务,赠送杨老师自写脚本进行结果数据后处理,课上讲授脚本编写思路和使用方法。
课程费用: 2980元。名额有限,欲报从速!提供增值税普通发票及邀请函。与MS电池计算专题课或MS光电热催化计算专题培训,同时报名优惠更多哦!
报名方式:识别下方二维码报名
,或者联系手机181-2638-7652。
可刷公务卡,请扫码填写报名信息以便我们提前为您准备发票等报销手续。
划重点:请先添加课程客服华算科技-妮妮微信报名再缴费!
版权申明