自由基不对称催化很难实现?这篇综述给你启发
2023-01-08
admin 38 0 核心提示:在我们的世界里,手性是最重要的基本属性之一。大至星系旋臂、大气气旋,小到矿物晶体以及如氨基酸和DNA等生命基础分子,都具
在我们的世界里,手性是最重要的基本属性之一。大至星系旋臂、大气气旋,小到矿物晶体以及如氨基酸和DNA等生命基础分子,都具有手性。与之相关,要开发能够改善人类生活的手性分子,通常需要对映选择性合成。过去20余年中,诺贝尔奖曾三次授予与不对称催化有关的领域:2001年的有机金属手性催化、2018年的酶定向进化(酶催化)以及2021年的不对称有机催化。这些策略最初是为了控制经典有机反应的立体选择性而开发,其反应机理涉及成对电子的流动。然而,近年来也出现了自由基化学相关的合成策略以解决合成化学中长期存在的挑战,并且尝试通过不对称催化来利用这些高活性的单电子物种。值得注意的是,如何控制这些短寿命、开壳中间体的生成及其立体选择性转化是长期存在的挑战,这也将继续推动自由基化学和不对称催化领域的不断创新。近日,美国
俄亥俄州立大学
的
David A Nagib
教授在
Chemical Reviews 对自由基化学中的不对称催化策略进行了规律性总结和展望。
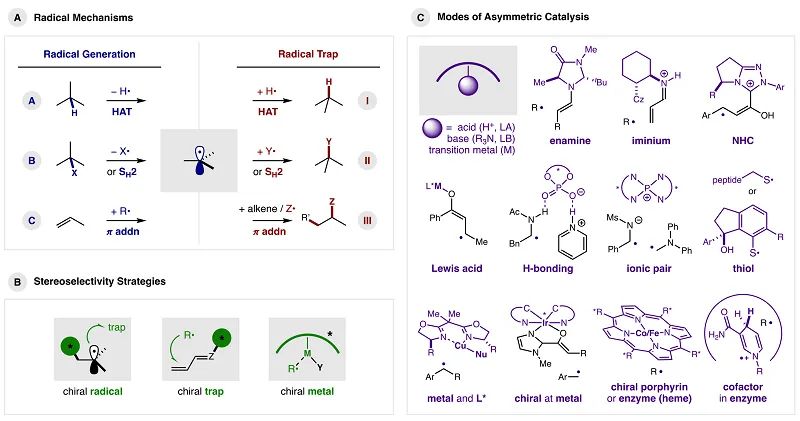
与传统的自由基产生方法(烷基锡、过氧化物、AIBN或紫外光)相比,光化学和电化学的发展可带来更多自由基参与的新颖且重要的转化。如图1A所示,自由基的产生一般分为以下三种模式:(A)通过氢原子转移(HAT)攫取H;(B)通过均裂攫取X基团(−X•)或均裂取代(
S
H S
H
图1. 自由基机理、立体选择性策略及不对称催化模式。图片来源:
Chem. Rev.
控制自由基立体化学最成功的方法包括在以下物种中引入手性(图1B):(i)自由基;(ii)自由基捕获剂;(iii)过渡金属(既可以产生自由基,也可以捕获自由基)。(i)将手性结合到反应的自由基组分上,这可能是对三种自由基捕获机理均适用且最理想的策略。当反应仅在手性催化剂结合时才能生成自由基时,可抑制外消旋背景反应。(ii)在自由基捕获剂上引入手性。该策略的价值在于各种催化策略的适用性,用于LUMO降低激活以促进亲核试剂的对映面、两电子加成。虽然通过这种活化(无论是催化上还是化学计量上)已经实现了自由基π-加成的高选择性,但尤需克服自由基化学中外消旋背景反应的固有挑战,这是因为开壳中间体即使没有催化活化也容易进行π-加成。最后,(iii)手性过渡金属具有通过手性配体(或手性金属配合物)对自由基和捕获剂进行立体化学控制的双重优势。一旦自由基与过渡金属结合,基本的有机金属机理即可控制自由基的终止过程(如非对映选择性还原消除)。
自由基化学中不对称催化的开创性工作以多种方式利用上述机理和策略进行手性控制。为了突出这些方法的显著特征,作者展示了各种反应模式的关键催化剂核心结构(图1C)。下面分别结合一些具有代表性和开创性的工作,对各种催化模式进行说明。
图2. SOMO活化的对映选择性有机催化。图片来源:
Science
手性胺可促进两种不同模式的不对称有机催化:烯胺和亚胺离子。前一种情况下,仲胺如咪唑啉二酮(源于氨基酸的环化;由MacMillan开发)或脯氨酸类似物(由List和Jorgensen开发)一直是最常见的。在这些情况下,手性胺和烯胺的活性π键之间的共价键确保在关键步骤中可接近手性环境。催化不对称烯胺活化的两种常见机理是:1)SOMO有机催化,其中烯胺的单电子氧化产生π-加成的自由基(由MacMillan和Sibi开发;
Science
,
2007
,
316
, 582;图2);2)光氧化还原有机催化,其中亲电自由基和亲核性烯胺结合(由MacMillan和Nicewicz开发;
Science
,
2008
,
322
, 77;图3),另外,Melchiorre发展了开壳亚胺离子活化(
Nature
,
2016
,
532
, 218;图4),其中亲核自由基与亲电亚胺离子结合(源自与α,β-不饱和醛的胺缩合)。而源于金鸡纳生物碱的简单伯胺可用于双电子亚胺催化,但氧化还原活性咔唑(Cz)部分的引入确保了自由基催化的立体控制,而不是发生未经活化的外消旋反应。
图3. 胺催化联合有机金属光氧化还原催化。图片来源:
Science
图4. 亚胺离子自由基捕获剂催化模式。图片来源:
Nature
另一类不对称有机催化是通过
N
-杂环卡宾(NHC)实现的。Rovis和池永贵课题组受到手性酶口袋内非手性硫胺素(维生素B1)极性反转反应性的启发,开创了控制自由基中间体的手性NHC催化策略(
J. Am. Chem. Soc
.,
2015
,
137
, 2416;图5)。与烯胺催化一样,这些仿生体系既可以使Breslow中间体的富电子π键进行自由基加成,也可以使该中间体反单电子氧化并与亲核试剂结合。
图5. 自由基中间体NHC催化。图片来源:
J. Am. Chem. Soc.
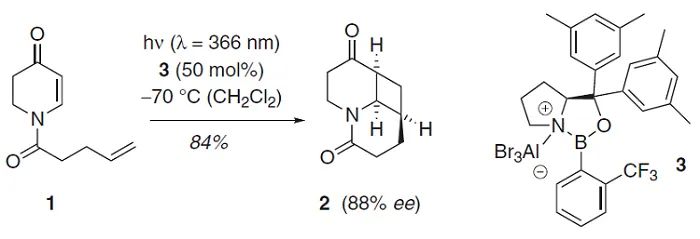
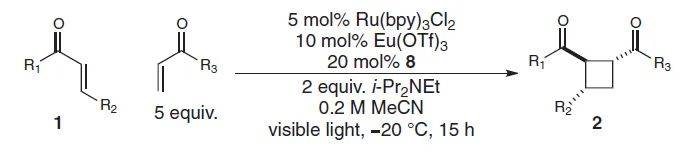
手性Lewis酸(LA)催化模式通常涉及与镧系金属(如:Eu、Gd)、金属Sc或非金属B的配合物,Bach(
Science
,
2013
,
342
, 840;图6)和Yoon(
Science
,
2014
,
344
, 392;图7)将其用于发展[2+2]光环加成反应。与传统的外消旋光化学不同,与LA配位的α,β-不饱和羰基可以在低能量可见光照射下产生自由基。两种机理包括通过能量转移(EnT)的三线态敏化和通过单电子转移(SET)的还原。在任何一种情况下,通过降低LA结合的羰基底物的三线态能量或还原电势,LA活化可选择性促进催化自由基的生成。类似地,手性Lewis酸Ti配合物也可介导环氧化物参与的不对称自由基转化,参见林松课题组的工作(
J. Am. Chem. Soc
.,
2018
,
140
, 3514,图8)。
图6. 手性LA催化烯酮分子内[2+2]光环加成。图片来源:
Science
图7. 手性LA催化不对称分子间[2+2]环加成。图片来源:
Science
图8. 手性Ti催化自由基接力[3+2]环加成。图片来源:
J. Am. Chem. Soc.
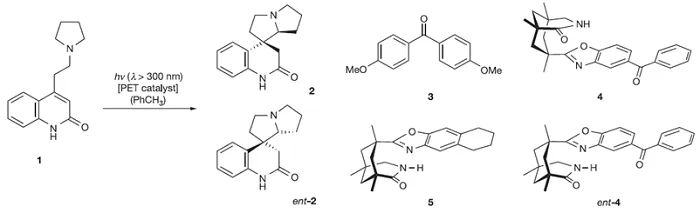
采用类似的原理,较弱的Brønsted酸也能活化底物,尽管通常是通过氢键相互作用。例如:Bach开发了与手性酰胺结合的光敏剂实现了[2+2]光环加成反应(
Nature
,
2005
,
436
, 1139,图9);Phipps使用手性磷酸(TRIP)催化剂实现了不对称Minisci加成反应(
Science
,
2018
,
360
, 419,图10)。
图9. 通过光诱导电子转移(PET)催化前手性化合物环化。图片来源:
Nature
图10. 手性磷酸催化杂环芳烃对映选择性的Minisci型加成。图片来源:
Science
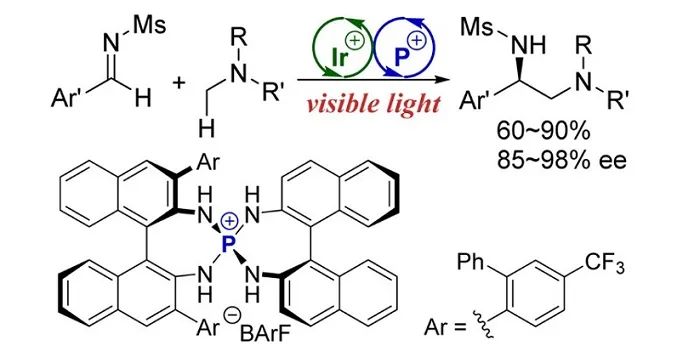
通过非共价相互作用,Ooi还在自由基化学中使用了离子对(
J. Am. Chem. Soc.
,
2015
,
137
, 13768;图11)。值得注意的是,尽管手性催化剂和一个自由基中间体仅由一个离子对连接在一起,但在两个α-氨基自由基的偶联中观察到高选择性。在这种情况下,一个膦盐与两个大的BINOL衍生胺(BINAM)绑定,从而将手性扩展到更大的空间。
图11. 经离子对中间体的不对称自由基转化。图片来源:
J. Am. Chem. Soc.
硫醇自由基介导的催化也是一种有效的策略。例如,Maruoka开发了一种手性硫酚,通过可逆巯基与烯烃的自由基加成实现[3+2]环化反应(
Nat. Chem
.,
2014
,
6
, 702;图12)。此外,Knowles和Miller通过将对映选择性自由基生成(通过SET氧化)和不同的不对称自由基捕获(通过硫醇介导的HAT)相结合,实现了α-脲的立体中心去消旋化(
Science
,
2019
,
366
, 364;图13)。
图12. 手性硫酚作为有机自由基催化剂。图片来源:
Nat. Chem.
图13. 激发态电子转移对环状脲的光致去消旋化反应。图片来源:
Science
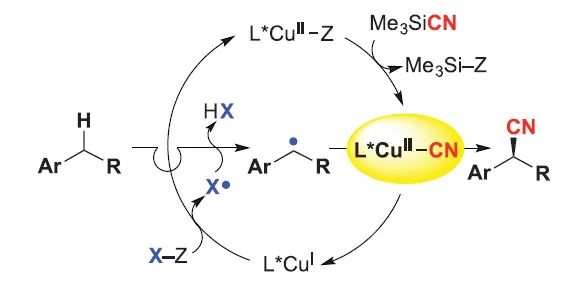
使用手性金属配合物来生成和捕获自由基中间体已通过多种策略得到证明。例如:刘国生课题组证实了手性铜配合物(通常使用双噁唑啉(Box)配体)是一种优秀的自由基捕获剂(
Science
,
2016
,
353
, 1014;图14),可通过C-C键形成捕获苄基自由基。该自由基可通过π-加成或HAT生成,后者可介导C-H键芳基化和氰化(
Acc. Chem. Res.
,
2018
,
51
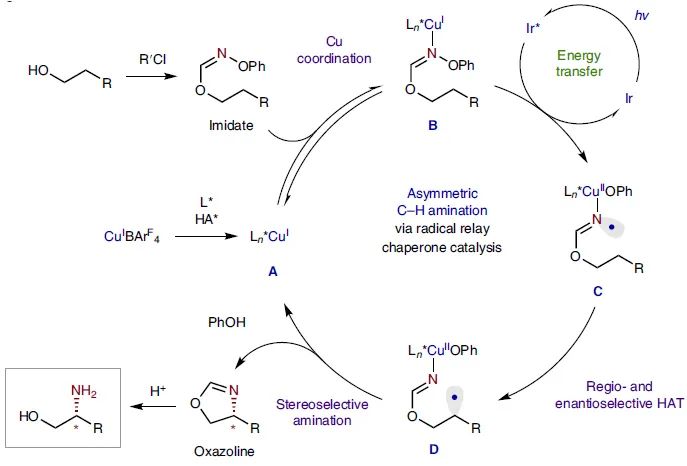
, 2036)。Nagib课题组将该方法进一步拓展,即通过imidate自由基介导的HAT偶联来实现C-H键胺化(
Nat. Chem
.,
2020
,
12
, 697;图15)。另外,刘心元课题组还利用手性磷酸盐组成的铜配合物捕获了烷基自由基(
Acc. Chem. Res
.,
2020
,
53
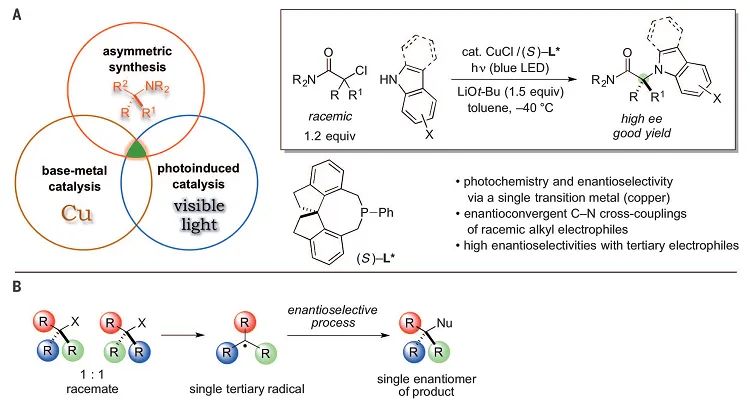
, 170);而Fu和Peters则是利用手性膦-铜配合物实现了C-N键交叉偶联反应(
Science
,
2016
,
351
, 681;图16)。
图14. Cu催化自由基接力对映选择性苄位C−H氰化。图片来源:
Science
图15. Cu催化对映选择性自由基C−H胺化。图片来源:
Nat. Chem.
图16. 手性膦-铜配合物催化C−N交叉偶联反应。图片来源:
Science
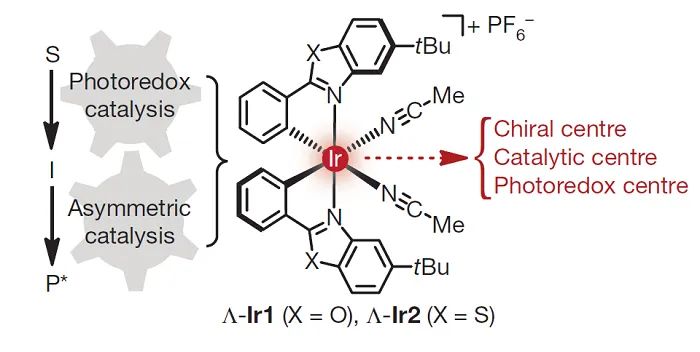
另外,Meggers还开发了金属Ir上具有手性的配合物(
Nature
,
2014
,
515
, 100;图17)。具体而言,Rh和Ir配合物可以与羰基结合形成手性自由基捕获剂。值得注意的是,这些配位不饱和的配合物仅在与底物络合时作为光敏剂选择性地引发自由基的形成。
图17. 用于不对称光氧化还原催化的手性Ir配合物。图片来源:
Nature
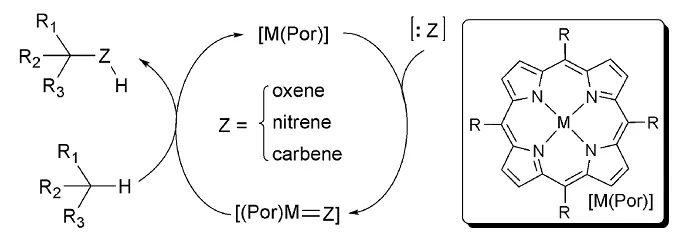
金属(Mn、Fe、Co)-卟啉在自由基化学和酶催化中具有很大的优势。氧化还原活性蛋白(细胞色素)中含铁辅因子(血红素)的存在对许多重要生物过程的化学机理至关重要,包括光合作用、电子传递和代谢C-H键氧化。Arnold已经证明这类酶工程可用于非自然反应,例如:卡宾和氮宾迁移反应(
Science
,
2013
,
339
, 307)。另外,Zhang通过将手性源直接引入到Co金属卟啉骨架上,证明了这种不对称反应也可以在没有酶口袋的情况下进行(
Chem. Soc. Rev.
,
2011
,
40
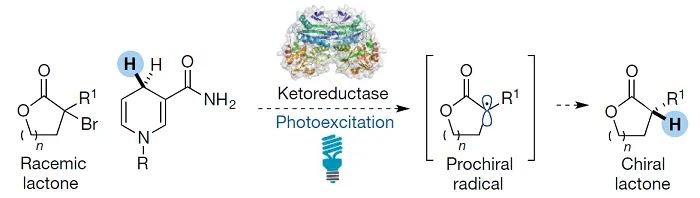
, 1899;图18)。其它酶促策略包括使用光激发还原酶进行还原和自由基环化。Hyster和Zhao使用烟酰胺(NADH)或黄素(FADH)辅酶因子并将其置于手性酶口袋中,实现了上述两类反应的不对称转化(
Nature
,
2016
,
540
, 414;图19)。尽管从细菌中分离出的野生型脱氢酶也可以实现这种转化,但工程酶通常具有更好的效率和选择性。
图18. 金属-卟啉催化C−H官能团化机理。图片来源:
Chem. Soc. Rev.
图19. 还原酶催化不对称自由基转化。图片来源:
Nature
目前,自由基不对称催化仍处于早期发展阶段。不过,化学家已经开发了一些开创性的策略,为长期存在的合成挑战提供了不错的解决方案。不对称自由基化学的未来发展前景光明,每一个化学工作者都可以发现有价值且新颖的催化不对称自由基转化。
Asymmetric Catalysis in Radical Chemistry
Chem. Rev
.,
2022
,
122
, 15989−15992, DOI: 10.1021/acs.chemrev.2c00622
https://www.x-mol.com/university/faculty/1644
点击“
阅读原文
”,查看
化学 • 材料
领域
所有收录期刊
版权申明