
点击蓝字关注我们
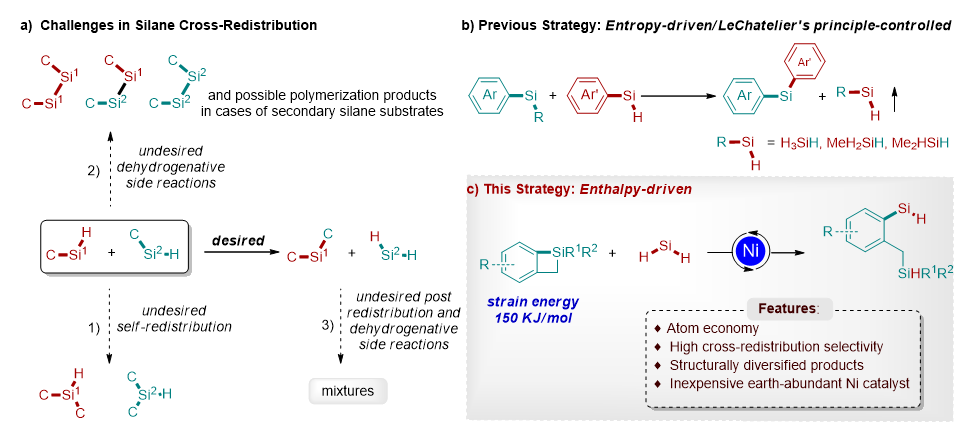
图 1: 硅烷再分配反应研究背景和本文的策略
经过一系列条件筛选,作者确定了最优条件为:当 Ni(COD) 2 的摩尔分数为 10 mol% , ( p -MeC 6 H 4 ) 3 P 的摩尔分数为 20 mol% 时,反应在 40 o C 下 3 h 内完成,以 90% 的产率得到所需的交叉再分配产物 3a ,并在此条件下对底物进行拓展。该反应具有广泛的底物范围,对于二芳基硅烷、一芳基一烷基硅烷、两烷基硅烷、三芳基硅烷、三烷基硅烷以及烷基芳基的三取代硅烷均能取得中等到良好的收率。其中包括含有供电子取代基、吸电子取代基和杂原子取代的硅烷底物。值得提出的是,对于工业上常用的硅氧烷和氯硅烷底物,也能在该条件下顺利反应得到双硅化合物。对于天然氨基酸衍生的复杂硅烷底物也能参与到反应中以中等收率得到相应的双硅产物(图 2 )。
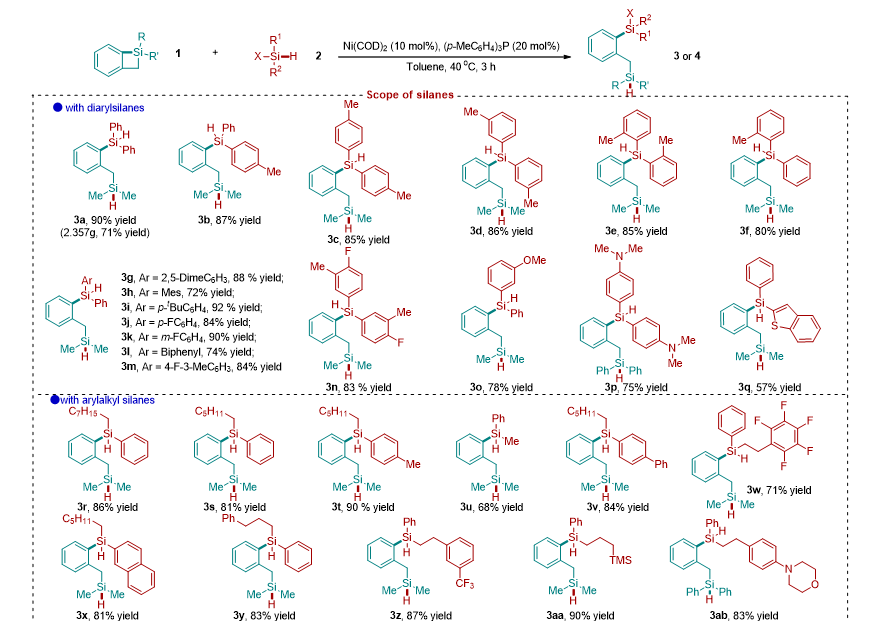
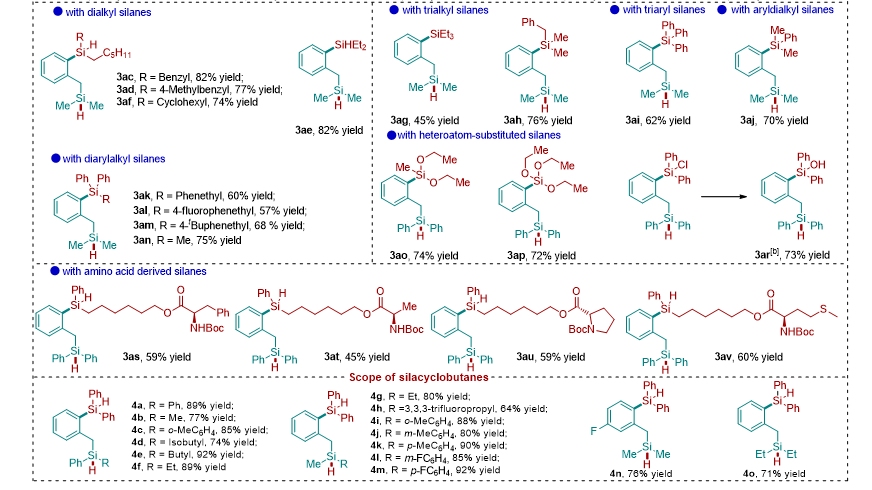
图 2: 底物拓展
随后作者尝试对含有硅氢的双硅化合物进行转化。通过 Rh 催化的分子内脱氢反应, 3a 以 81% 的产率转化为环双硅化合物 5 。除了过渡金属催化剂外, B(C 6 F 5 ) 3 在硅氢化反应中也表现出良好的性能。作者以 5% 的 B(C 6 F 5 ) 3 为催化剂,实现了丙酮的硅氢化反应,合成了六元环硅氧烷 6 ,产率 84% 。在类似条件下,苯甲腈经历可能的中间体 7 经水解后,以 84% 的产率得到苯甲胺 8 。当硅上取代基的电性不同时,硅氢的反应活性是不同的,作者利用这一特性实现了在同一个分子中有两个硅氢键的选择性硅氢化,在 B(C 6 F 5 ) 3 的催化下 4b 能与苯乙烯反应,选择性的在烷基取代的硅氢键上发生硅氢化以 78% 的收率得到化合物 9 。剩余的一个硅氢也可以在碱性条件下高效的被转化为硅羟基,以 81% 的收率得到化合物 10 。通过 C-H 硅烷化反应合成六元硅杂环化合物更具挑战性,因为需要生成高能量的七元环金属中间体。作者发现了一种 Rh 催化分子内碳氢键硅基化反应,以硅烷再分配产物为底物来实现此类分子的构建。结果表明,以 [Rh(COD)Cl] 2 为催化剂, Xantphos 为配体,在甲苯中反应效果较好,以 60% ~ 80% 的产率合成了 11a 到 11h 的二硅苯并环己烷 ( 图 3) 。这些后续化学转化都展示了作者开发的方法学的合成应用价值。

图 3: 双硅 产物的转化与应用
为了更加深入的了解硅烷与环硅化合物的再分配反应的机理,作者进行了一系列的机理验证实验,并结合 DFT 计算,对这个反应的机理进行研究。首先,为了探究反应是否经历自由基的历程,作者在标准模板反应中加入自由基抑制剂 TEMPO ,反应可以进行,这表明产物的形成不太可能涉及自由基机制 ( 图 4-a) 。在模板反应中,作者将 Ph 2 SiH 2 替换为 Ph 2 SiD 2 ,发现产物中的硅氢均是氘代的硅氢,说明产物中的氢来源于硅烷 ( 图 4-b) 。当将 Ph 2 SiH 2 /Ph 2 SiD 2 (1 : 1) 混合加入到模板反应中时, KIE 只有 1.08 左右,并没有发现大的 KIE 效应 ( 图 4-c) 。为了更清楚的了解硅氢键的断裂过程,作者还进行了平行实验进行 KIE 的探究,得到 KIE 的数值为 1( 图 4-d) ,说明在这个反应中硅氢键的断裂在动力学上并不显著,也就是说硅氢键的断裂并没有参与到决速步中。作者对反应组分的动力学进行了研究 ( 图 4-e) 。作者首先研究了 Ph 2 SiH 2 不同浓度下的初始速率,发现该速率对 Ph 2 SiH 2 的浓度呈现零级动力学效应,这与 KIE 结果一致,表明 Ph 2 SiH 2 的 Si-H 键的断裂不参与到决速步中。然后作者接着研究了催化剂和 1a 不同浓度下的初始速率,发现反应速率与催化剂( Ni(COD) 2 +2( p -MeC 6 H 4 ) 3 P )和 1a 的浓度呈一级动力学效应,表明 1a 在催化剂作用下 C-Si 键的断裂可能是反应的决速步骤。
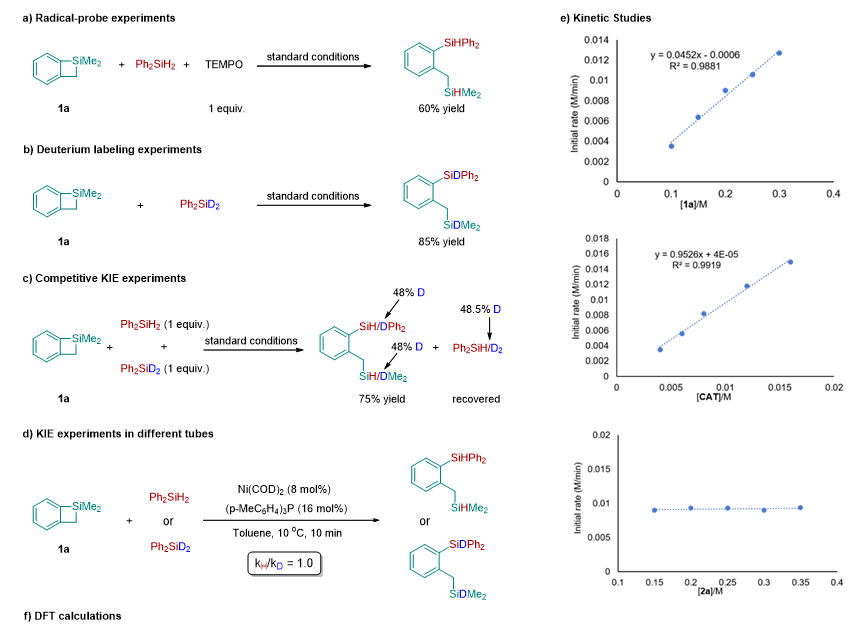
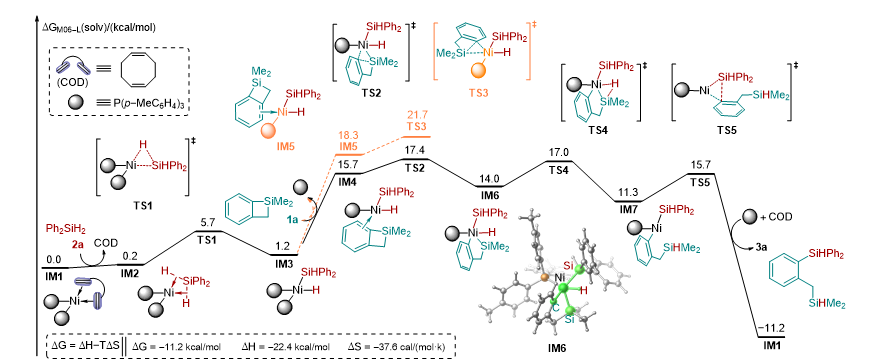
图 4: 机理研究与 DFT 计算
基于这些实验结果和有关镍催化反应的相关报道 , 作者提出了一个可能的镍催化二苯基硅烷和苯并四元环硅的交叉再分配反应的催化循环机理。如图 5 所示,首先 Ni(COD) 2 和 2( p -MeC 6 H 4 ) 3 P 络合配位生成零价镍物种 I 与 Ph 2 SiH 2 发生配体交换得到中间体 II , 然后 Ni(0) 与二苯基硅烷的硅氢键发生氧化加成生成二价镍中间体 III . 随后苯并环硅的硅碳键与二价镍中间体 III 发生氧化加成得到四价镍中间体 I V 。最后,硅氢键与硅碳键分别发生还原消除得到目标产物 3a 并重生了催化剂活性物种 I 。 DFT 计算的结果进一步支持了作者提出催化循环机理,反应整个历程的吉布斯自由势能如图 4-f 所示。 Ni(0) 中间体 IM1 与二苯基硅烷 2a 发生配体交换生成中间体 IM2 所需要的能量仅为 0.2 kcal/mol 。随后硅氢键的氧化加成经过过渡态 TS1 生成 Ni(II)- 中间体 IM3 所需要的的能量为 5.5 kcal/mol 。这个计算结果表明零价镍可以很容易的被二苯基硅烷氧化为二价镍。另一种硅烷底物苯并四元环硅 1a 可以通过配体交换与 Ni(II) 中心配位,后续发生的配体交换如果发生在镍上氢原子的反式会生成中间体 IM5 ,相应的通过过渡态 TS3 的 Si-C 键氧化加成反应需要 21.7 kcal/mol 的活化自由能。当配体交换发生在镍上硅原子的反式会形成中间体 IM4 ,相应的通过过渡态 TS2 的 Si-C 键氧化加成反应生成 Ni(IV) 中间体 IM6 需要 17.4 kcal/mol 的活化自由能,比经历 TS3 所需要的能量低 4.3 kcal/mol 。随后的 Si-H 还原消除通过过渡态 TS4 生成 Ni(II) 中间体 IM7 ,势垒为 2.7 kcal/mol 。最后,通过过渡态 TS5 的 Si-C 键还原消除得到产物 3a 并再生 Ni(0) 物种 IM1 。该步骤的势能垒为 4.4 kcal/mol ,自由能降低为 22.5 kcal/mol 。计算结果表明,通过 Si-C 键氧化加成生成的 Ni(IV) 络合物具有最高的活化自由能,可能是决速步。计算的结果与动力学结果是一致的。
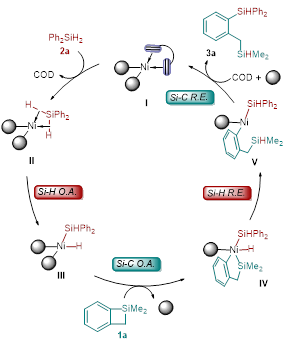
图5: 机理预测
在该工作中,沈晓课题组报道了一种硅烷和苯并硅杂环丁烷在镍催化下的选择性交叉再分配反应。该反应能够成功的关键是由于在熵驱动下苯并四元环硅环张力的释放。这与之前的通过生成硅烷气体的焓驱动的硅烷再分配反应在反应模式上是不一样的。 该反应具有广泛的底物范围。 实验和计算相结合的机理研究支持反应是通过Ni(0)-Ni(II)-Ni(IV)催化循环进行的。其中环硅通过环张力释放的氧化加成步骤是整个再分配反应能够成功的关键 。
高等研究院博士研究生陈绍维为第一作者。计算部分的工作由重庆大学博士研究生何晓倩完成。武汉大学沈晓教授、重庆大学蓝宇教授和加州大学洛杉矶分校Houk教授为通讯作者。
该研究得到了中国国家自然科学基金委、中国中央高校基本科研业务费、武汉大学人才启动经费和美国国家自然科学基金委的支持。
作者简介
高等研究院博士研究生陈绍维为第一作者。计算部分的工作由重庆大学博士研究生何晓倩完成。武汉大学沈晓教授、重庆大学蓝宇教授和加州大学洛杉矶分校Houk教授为通讯作者。
沈晓课题组常年招收博士后 , 研究生,感兴趣的同学请直接联系沈晓老师( xiaoshen@whu.edu )
原文链接
https://onlinelibrary.wiley.com/doi/10.1002/anie.202213431
相关进展
武大沈晓教授课题组 Angew: α-三氟甲基取代烯烃与环丙醇的光催化串联反应合成稠合偕二氟氧杂环丁烷
武汉大学沈晓教授团队Angew:三(二)氟甲基酰基硅衍生的卡宾与烯烃的非对映选择性环丙烷化反应
化学与材料科学原创文章。 欢迎个人转发和分享,刊物或媒体如需转载,请联系邮箱: chen@chemshow.cn

扫二维码|关注我们
微信号 : Chem-MSE
欢迎专家学者提供化学化工、材料科学与工程产学研方面的稿件至chen@chemshow.cn,并请注明详细联系信息。化学与材料科学®会及时选用推送。
